Página anterior « Indice del Curso »Siguiente Pagina
Análisis de datos NGS

CONTENIDO
SIMULACION DE READS
SimRead -- Program to create simulation reads for illumina sequencing in GNU/Linux.
created by Francisco Ascue (francisco.ascue131@gmail.com)
usage: SimRead -n <project> -g <N.A. NCBI> -r <20000> -f <fastqfile>
where:
-h Show this help text
-n <name> Name of project
-g <N.A. NCBI> N.A. of reference genome
-r <number> Number of reads simulated
-f <str> Name of fastq files
-s <number> set the seed value (default: 42)
-m <float> Rate of mutation of reference genome
-e <float> Rate of error sequencing simulation
SimRead -n SARS -g NC_045512.2 -r 6000 -f sars2 -e 0.01
── $SARS/
│ └── data/ <- Folder to store reads and references files
│ ├── reads/ <- Reads illumina simulated
│ ├──sars2_1.fastq <- Forward read
│ ├──sars2_2.fastq <- Reverse read
│
│ ├── reference/ <- Host genomes files (.fasta)
│ ├──NC_045512.2.fasta <- NCBI download fasta file
│
│ └── results/ <- Folder to store data generated during processing steps
│
│ └── scripts/ <- Folder to store scripts for data processing
├── logs/ <- Results logs during processing steps
CONTROL CALIDAD
fastqc -t 2 cavtsc_forward_paired.fq.gz cavtsc_reverse_paired.fq.gz -o /mnt/disco2/fascue/cporcellus/results/fastqc/
FILTRADO DE READS
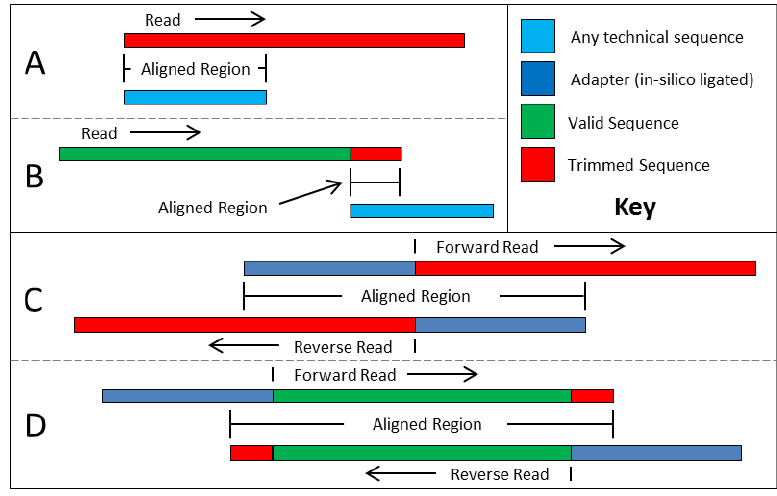
TrimmomaticPE -phred33 -threads 2 file_1.fastq file_2.fastq file_forward_paired.fq.gz file_forward_unpaired.fq.gz file_reverse_paired.fq.gz file_revers_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50
ILLUMINACLIP:<fastaWithAdaptersEtc>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>
LEADING:<quality>
TRAILING:<quality>
SLIDINGWINDOW:<windowSize>:<requiredQuality>
MINLEN:<length>
ALINEAMIENTO


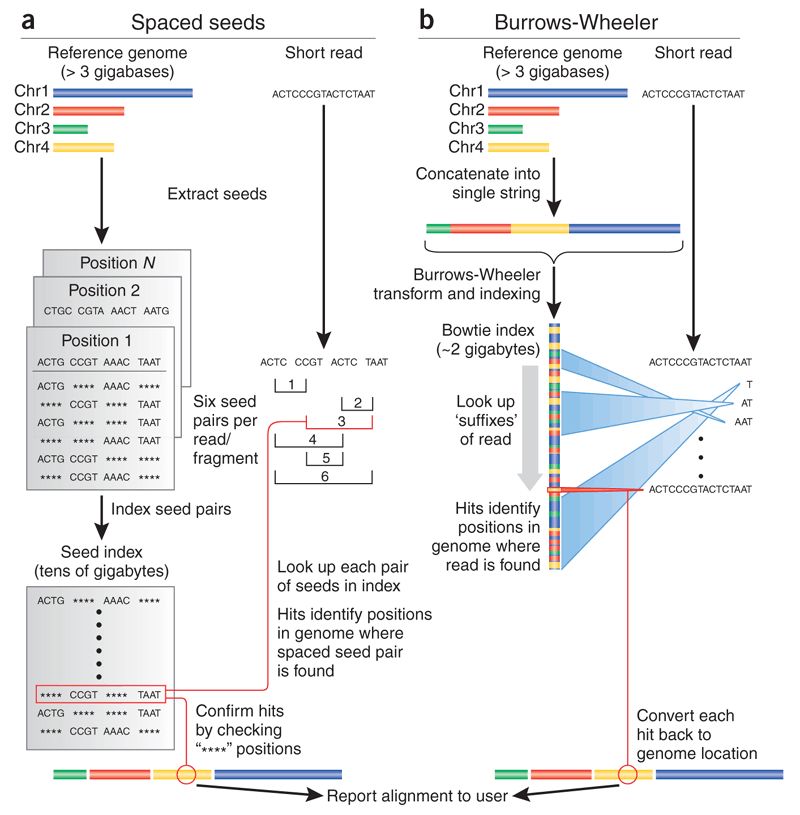
Preparacion del index
#!/bin/bash
###bowtie2-build
###CONSTANTS
WD="~/Curso_transcriptomica/SARS"
REF="${WD}/data/reference/NC_000.fasta"
IDX="${REF}/index"
###EXECUTION
echo "started at ´date´"
echo "mkdir -p ${IDX}"
mkdir -p ${IDX}
bowtie2-build -threads 2 ${REF} ${IDX}/sars
echo "Finished at ´date´"
Alineamiento de secuencias
#!/bin/bash
###bowtie2
###CONSTANTS
WD="~/Curso_transcriptomica/SARS"
REF="${WD}/data/reference/NC_000.fasta"
RES="${WD}/results"
READS="${WD}/data/reads/"
r1="${READS}/sars2_1.fq"
r2="${READS}/sars2_2.fq"
OD="${RES}/map"
###EXECUTION
echo "started at `date`"
echo "mkdir -p ${OD}"
mkdir -p ${OD}
bowtie -end-to-end -I 0 -X 1000 -p 30 -x ${REF} -1 $r1 -2$r2 -S ${OD}/cavtsc.sam
samtools view -u@ 8 ${OD}/cavtsc.sam | samtools sort -@ 40 -o ${OD}/cavtsc.sorted.bam -
samtools index ${OD}/cavtsc.sorted.bam
echo "Finished at `date`"
ENSAMBLAJE



spades.py -1 file_1.fq -2 file_2.fq -s file.single.fq -m 2 -k 31,41,51 -o outputDir
#!/bin/bash
### CONSTANTS
READS="${MNTD3}/data/reads"
RES="${MNTD3}/results/maps"
OD="${RES}/assembly"
WD="~/Curso_transcriptomica/SARS"
REF="${WD}/data/reference/NC_000.fasta"
RES="${WD}/results"
READS="${WD}/data/reads/"
r1="${READS}/sars2_1.fq"
r2="${READS}/sars2_2.fq"
OD="${RES}/map"
### EXECUTION
echo "Started at `date`"
for i in SA42911 SA42912 SA42913 SA42914 SA42976 SA42977 SA42978 SA42979 SA42980 SA42981
do
mkdir -p ${OD}/$i/spades
echo "spades.py -1 ${RES}/${i}map/mito${i}_1.fq -2 ${RES}/${i}map/mito${i}_2.fq -m 4 -t 4 -k 41,51,61 -o ${OD}/${i}/spades"
spades.py -1 ${RES}/${i}map/mito${i}_1.fq -2 ${RES}/${i}map/mito${i}_2.fq -m 4 -t 4 -k 41,51,61 -o ${OD}/${i}/spades
done
echo "Finished at `date`"
VALIDACION DE ENSAMBLAJE
quast.py -r refseq.fasta -e -o outputdir fasta_assemblyScaffold.fa