Página anterior « Indice del Curso »Siguiente Pagina

BASH SCRIPT PARA NGS
CONTENIDO
Pipelines y Scripts para Bioinformática
BASH PIPELINES

Conteo de secuencias en FastQ
fgrep -i "@S" file.fq | wc -l
cat file.fq | echo $((`wc -l`/4))
Nombre de los genes anotados
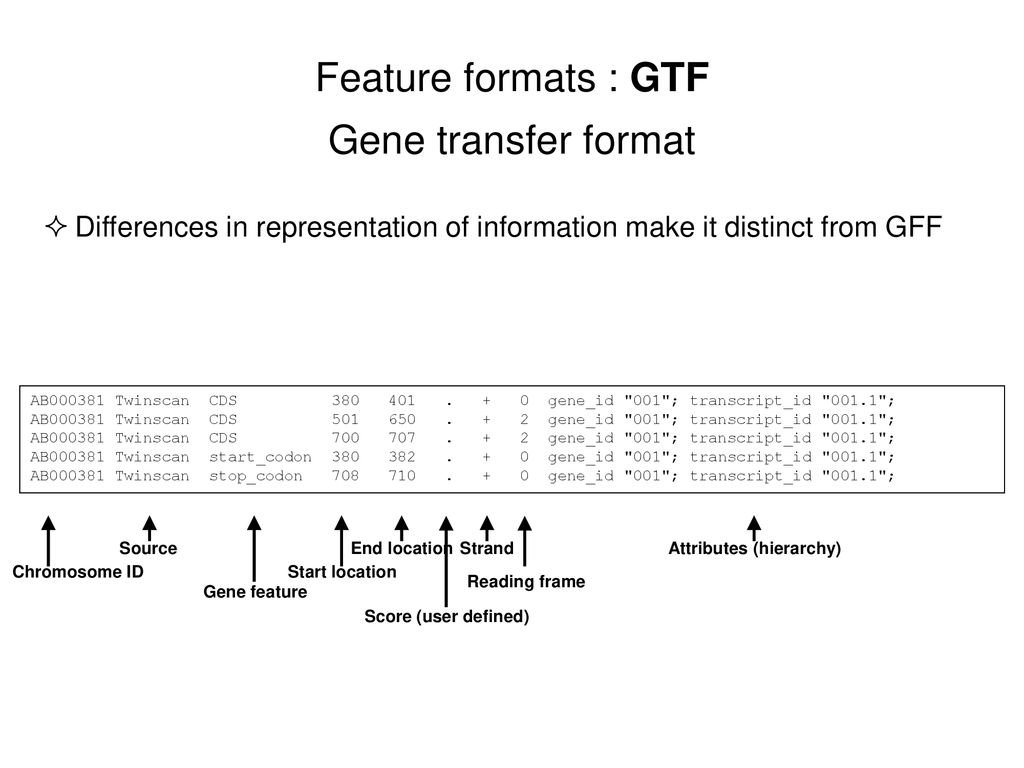
grep $'\tgene\t' sequence.gff3 | perl -ne '/ID=([^;]+)/ and printf("%s\n", $1)'
grep $'\tgene\t' sequence.gff3 | awk '{print $9}' | cut -d';' -f1 | sed "s/ID=//g"
Mesclando scripts:
grep $'\tgene\t' sequence.gff3 | awk '{print $5-$4";"$9}'| sed 's/Name=//g' | awk -F';' '{print $3"\t"$1}' | sort -k2n > Tamaño_genes.txt
awk '{print $3}' sequence.gff3 | sort -d | uniq -c
BASH SCRIPTS
she bang
#!interpreter [optional-arg]
#!/bin/sh
#!/bin/bash
#!/usr/bin/python3
#!/usr/bin/awk -f
#!/bin/bash
##CONSTANTES
VAR1="UUU"
VAR2="UUC"
##EJECUCION
for i in {A,U,G,C}{A,U,G,C}{A,U,G,C}
do
if [ "$i" = "$VAR1" ] || [ "$i" = "$VAR2" ]; then
echo "$i Phe"
fi
done
Permisos
# cambia permisos a archivos y carpetas
# d rwx rwx rwx / (propietario) (grupos) (demas)
# r 4
# w 2
# x 1
chmod 775 ACGT.bash
chmod +rwx ACGT.bash
# R recursivo
chmod -R 755
Correr un script
<lenguaje> script.ext
### Sin permisos
bash ACGT.bash
### Con permisos
./ACGT.bash
### Segundo plano
./ACGT.bash&
Correr en segundo plano o screen
# Añadir el caracter &
bash scrip.bash&
### Activando un screen
screen -S name
./ACGT.bash
## salir del screen CTRL A + D
## ingresar al screen nuevamente
scren -ls
screen -r name
Detener procesos
# muestrar los procesos actuales
ps
pstree
top -u user
#detener procesos por comando
kill PID
top -u user
# presiona "k" y indica el PID
htop
# buscar el proceso y presionar "k"
SYNTAXIS
#!/bin/bash
###COMENTARIOS
###VARIABLES (numericos (enteros o flotantes), strings (""), booleanos (T / F)
VAR1=""
VAR2=""
VAR3=""
###EXECUTION ( se puede ejecutar comandos que se usan en la terminal )
echo "started at ´date´" ## ´date´ (permite ejecutar comandos dentro de un print
mkdir -p ${VAR} ## puede intercambiar variables para usarlos dentro de los comandos.
### sintaxis de comparacion
== is equal to ; if [ "$a" == "$b" ]
!= is not equal to ; if [ "$a" != "$b" ]
< is less than; if [[ "$a" < "$b" ]]
< is greater than if [[ "$a" > "$b" ]]
AND (&&) y OR(||)
-eq is equal to if [ "$a" -eq "$b" ]
-ne is no equal to if [ "$a" -ne "$b" ]
-gt is greater then if [ "$a" -gt "$b" ]
-ge is greater then or egual to if [ "$a" -ge "$b" ]
-lt is less than if [ "$a" -lt "$b" ]
-le is less than or equal to if [ "$a" -le "$b" ]
### estructura basica de una condicional if
if [ "$i" != "UGA" ]; then
echo "$i phe"
fi
### estructura basica de un while
while [ number -gt 4 ]; do
echo "loop1"
echo "loop2"
echo "loop3"
done
### estrucutura basica de un for
for i in a b c
do
comand <options> target
done
Introducción a datos NGS
ARCHIVOS PARA DATOS NGS
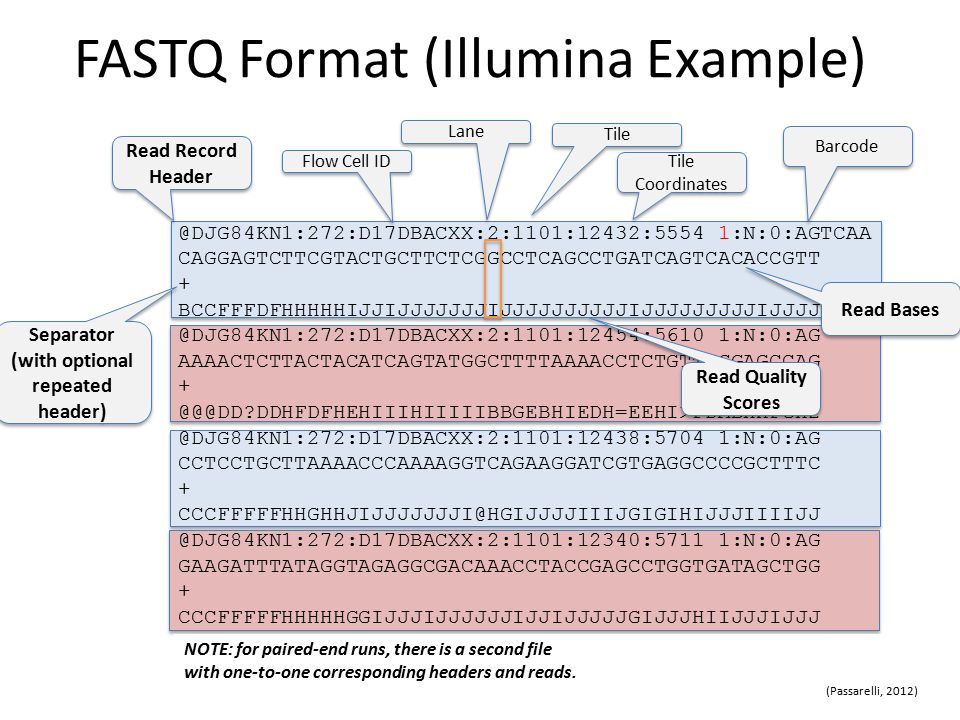
FASTQ y SAM (Sequence Alignment Map)

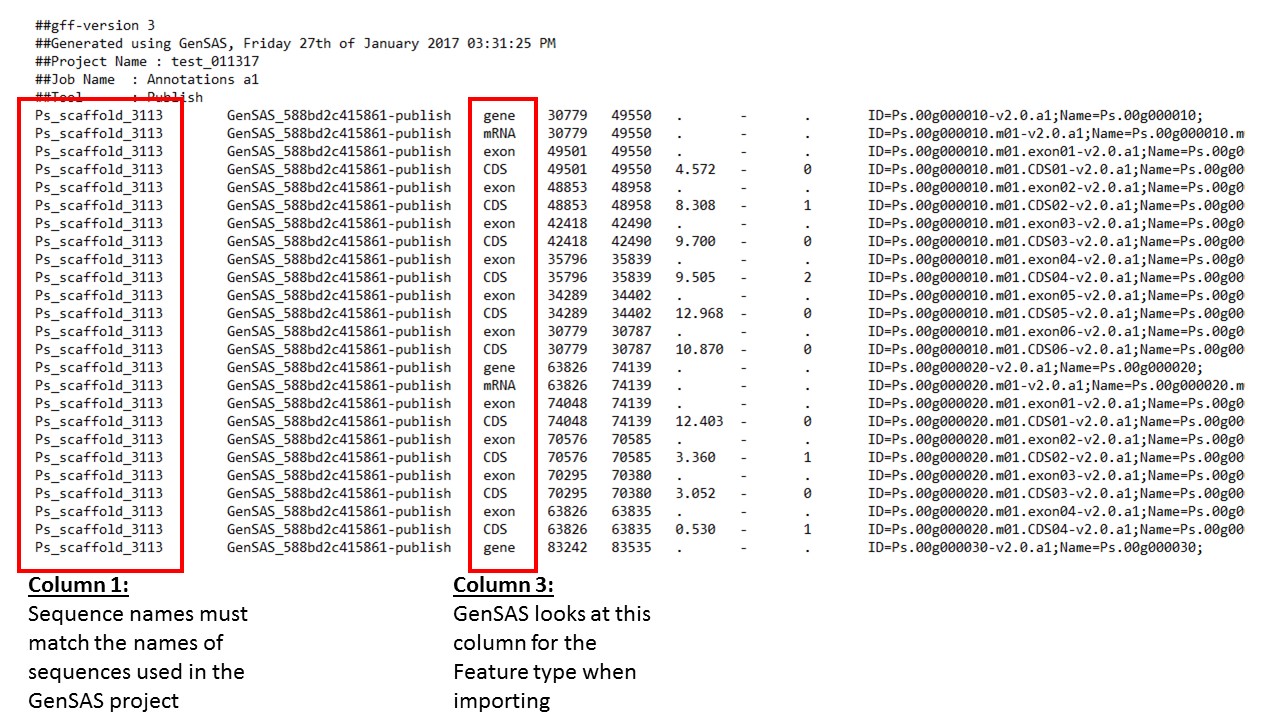
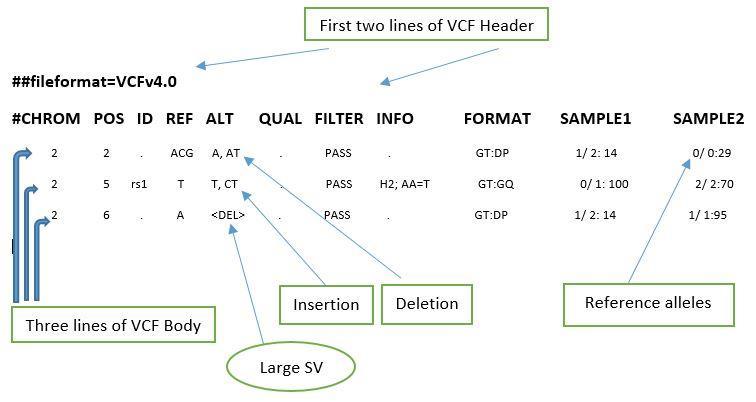
FORMATOS DE ANOTACION(GFF(general feactures format), GTF(gene transfer format), VCF)