Página anterior« Indice del Curso »Siguiente Página
Analisis de datos RNA-Seq
Introducción
Instalacion de programas
git clone https://github.com/FranciscoAscue/Curso_transcriptomica.git
#Ubuntu20
bash preinstall.sh
#Ubuntu18
bash preinstall.sh
Directorio de trabajo
── RNA_Seq/
│ └── genome/ <- genoma de referencia (.FASTA) , anotación de genoma (.GTF/.GFF)
│
│ └── reads/ <- RNAseq data
│
│ └── results/ <- Archivos generados
│ ├── quality/ <- Archivos de calidad
│ ├──multiQC/ <- calidad conjunta
│ ├──rawdata/ <- calidad de data cruda
│ ├── trimmed/ <- Archivos filtrados
│ ├── sortmerna/ <- Archivos filtrados de rRNA
│ ├── aligned/ <- Secuencias alineadas a rRNA databases (con contenido de rRNA)
│ ├── filtered/ <- Secuencias con rRNA removidos (libre de rRNA)
│ ├── logs/ <- logs
│ ├── map/ <- Alineamientos al genoma de referencia
│ ├── aligned_bam/ <- Archivos de alineamiento (.BAM)
│ ├── aligned_logs/ <- logs
│ ├── counts/ <- Conteo de secuencias finales
│
│ └── sortmerna_db/ <- rRNA databases
│ ├── rRNA_databases/ <- rRNA (bacteria, archea y eukaryotes)
│
│ └── scripts/ <- Scripts usados con el curso
REPOSITORIO DE DATOS DE TRANSCRIPTOMA
Los datos utilizados en este curso fueron descargados de EMBL-EBI, usando la base de datos de Expression Atlas. Para mas detalles del los datos utilizados en este curso puede revisar el siguiente Link o puede visualizar directamente a partir de la base de datos Expression Atlas E-GEOD-46817
CONTENIDO
- DESCARGAR SECUENCIAS
- CONTROL DE CALIDAD
- FILTRADO DE SECUENCIAS
- FILTRADO DE rRNA
- ALINEAMIENTO
- CONTEO DE SECUENCIAS
- IMPORTAR A R
DESCARGAR SECUENCIAS
Descargar desde SRA-db
### Sra-tool kit
prefetch -O ~/Minicurso_transcriptomica/RNA_seq/reads SRR8495--
# prefetch.2 -a 'ascp|/PATH_to_aspera/.aspera/connect/etc/asperaweb_id_dsa.openssh' -X 100G -O SRR8495-- SRX8495--
fasterq-dump SRR8495--.sra -S -m 1G -O ~/Minicurso_transcriptomica/RNA_seq/reads -e 2
Para descargar datos utilizados en el curso
wget --load-cookies /tmp/cookies.txt "https://docs.google.com/uc?export=download&confirm=$(wget --quiet --save-cookies /tmp/cookies.txt --keep-session-cookies --no-check-certificate 'https://docs.google.com/uc?export=download&id=FILEID' -O- | sed -rn 's/.*confirm=([0-9A-Za-z_]+).*/\1\n/p')&id=FILEID" -O FILENAME && rm -rf /tmp/cookies.txt
| SAMPLE | FILEID | FILENAME |
|---|---|---|
| SRR849504 | 1p6jTh3JYK3mCmusOj3dj_fRLc2G4v5-s |
NC_000021.9_1.fq |
1R9R9d9vETQNuOwKOqXUQfkIufmrBXWef |
NC_000021.9_2.fq | |
| SRR849505 | 1dGt-SSyze4yuNqtN9MPXGIeBj42I3YFf |
NC_000021.9_1.fq |
18eSbO-rEWt9iv22vlPlb8YaisudC8UIq |
NC_000021.9_2.fq | |
| SRR849506 | 1FGk-0jAOMk2msL6buFAnMhYgpXve4Z8t |
NC_000021.9_1.fq |
18vp7LD-makAdcrpyaoqNOp6D1J6dvtEr |
NC_000021.9_2.fq | |
| SRR849507 | 1WceQz3qDvlFAhBQMt4YJPSCguUYNcaJr |
NC_000021.9_1.fq |
1CGjXGrTS6iIJ2e_Y2bcvfBI60FenlAB7 |
NC_000021.9_2.fq | |
| SRR849508 | 1HZBUwCGjuYFjyHkavdzkh6rQ1W3EFA9l |
NC_000021.9_1.fq |
1pTT17NMb2UYs1ZRS96a2z8nXyDEz2_tR |
NC_000021.9_2.fq | |
| SRR849509 | 1_1s_6A-sRxhcWgIUTMU3PiCgp6gbEC17 |
NC_000021.9_1.fq |
1ZetrrsOnbHlGLD43RwwJjBSBVP35ABy- |
NC_000021.9_2.fq | |
| SRR849510 | 1XC8kvuDazTsf5_GDLGoE-c4WrRfZyagw |
NC_000021.9_1.fq |
1Hb9PfckXKXT_pegNF_AOA3SVa4oh6zRA |
NC_000021.9_2.fq |
CONTROL DE CALIDAD
# fastqc
fastqc -t 2 NC_000021.9_1.fq NC_000021.9_2.fq -o ~/Minicurso_transcriptomica/results/quality/multiQC/
#### Output
── results/multiQC/
└── NC_000021.9_1.html <- Estadisticas de calidad NC_000021.9_1.fq
└── NC_000021.9_2.html <- Estadisticas de calidad NC_000021.9_2.fq
└── NC_000021.9_1.zip <- Datos obtenidos por Fastqc
└── NC_000021.9_2.zip <- Datos obtenidos por Fastqc
# multiqc
cd ~/Minicurso_transcriptomica/results/quality/multiQC/
multiqc .
#### Output
── results/multiQC/
└── multiqc_report.html <- Estadisticas de calidad de todos los archivos fastqc (.html)
└── multiqc_data/ <- Datos que multiqc encontró de varios archivos de registro (.zip)
FILTRADO DE SECUENCIAS
#trim_galore
trim_galore --quality 20 --fastqc --length 25 --output_dir results/trimmed/ reads/SRR849504/NC_000021.9_1_trimmed.fastq
FILTRADO DE rRNA
wget -P sortmerna_db https://github.com/biocore/sortmerna/archive/2.1b.zip
## sortmerna
# Guardar en una variable los rRNA databases
sortmernaREF=sortmerna_db/rRNA_databases/silva-arc-16s-id95.fasta,sortmerna_db/index/silva-arc-16s-id95:\
sortmerna_db/rRNA_databases/silva-arc-23s-id98.fasta,sortmerna_db/index/silva-arc-23s-id98:\
sortmerna_db/rRNA_databases/silva-bac-16s-id90.fasta,sortmerna_db/index/silva-bac-16s-id95:\
sortmerna_db/rRNA_databases/silva-bac-23s-id98.fasta,sortmerna_db/index/silva-bac-23s-id98:\
sortmerna_db/rRNA_databases/silva-euk-18s-id95.fasta,sortmerna_db/index/silva-euk-18s-id95:\
sortmerna_db/rRNA_databases/silva-euk-28s-id98.fasta,sortmerna_db/index/silva-euk-28s-id98
sortmernaREF=sortmerna_db/rRNA_databases/silva-arc-16s-id95.fasta,sortmerna_db/index/silva-arc-16s-id95:\
sortmerna_db/rRNA_databases/silva-arc-23s-id98.fasta,sortmerna_db/index/silva-arc-23s-id98:\
sortmerna_db/rRNA_databases/silva-bac-16s-id90.fasta,sortmerna_db/index/silva-bac-16s-id95:\
sortmerna_db/rRNA_databases/silva-bac-23s-id98.fasta,sortmerna_db/index/silva-bac-23s-id98:\
sortmerna_db/rRNA_databases/silva-euk-18s-id95.fasta,sortmerna_db/index/silva-euk-18s-id95:\
sortmerna_db/rRNA_databases/silva-euk-28s-id98.fasta,sortmerna_db/index/silva-euk-28s-id98
# Run SortMeRNA (~4min)
sortmerna --ref $sortmernaREF --reads results/trimmed/NC_000021.9_1_trimmed.fq \
--aligned results/sortmerna/aligned/sample_aligned.fq \
--other results/sortmerna/filtered/sample_filtered.fq \
--fastx \
--log \
-a 4 \
-v
# mover los log a resultados
mv -v results/
ALINEAMIENTO
Es puede tomar con los datos completos alrededor de 30 minutos
Para el curso utilizaremos unicamente el cromosoma 21 NC_000021.9
Antes de empezar con el alineamiento se va desargar el archivo de anotación gff3 del cromosoma 21 y se utilizará el programa gffread para convertir al formato gtf, para mas detalles revisar el repositorio del programa aqui
## Instalacion a travez de apt
sudo apt install gffread
## Instalacion desde respositorio.
cd /some/build/dir
git clone https://github.com/gpertea/gffread
git clone https://github.com/gpertea/gclib.git
cd gffread
make release
Indexar el genoma con gtf-file
Descargamos primero el archivo gff3 del NCBI
wget -O NC_000021.9.gff3 "https://www.ncbi.nlm.nih.gov/sviewer/viewer.cgi?db=nuccore&report=gff3&id=NC_000021.9,NC_000021.9"
Convertimos el archivo gff3 a gtf
## instalación
sudo apt install gffread
gffread -E -O -T NC_000021.9.gff -o NC_000021.9.gtf
Indexar el genoma con STAR
| Parámetro | Descripción |
|---|---|
--runMode |
indica el tipo de opcion que utilizará STAR, en este caso queremos generar un índice del genoma por lo que utilizamos la flag genomeGenerate |
--genomeDir |
indica donde se guardaran los resultados del indice y la ubicación de los archivos del genoma |
--genomeFastaFiles |
indica donde estan almacenadas las secuencias del genoma en formato FASTA |
--sjdbGTFfile |
sj: splice junction db: database GTFfile: archivo GTF indica la ubicación del archivo GTF para mejorar e improvisar el mapeo dado el modelo de los genes |
--sjdbOverhang |
Especifica el largo a considerar de la secuencia genómica alrededor del splice junction, este valor esta ligado al largo de los reads y deberia ser max(ReadLength) - 1 |
--runThreadN |
total de hebras que se ejecutaran en paralelo, este número no debe sobrepasar la cantidad de cores que tiene un computador y pruebas de escalamiento deberian ser ejecutadas para calcular el óptimo |
STAR \
--runMode genomeGenerate \
--genomeDir genome/star_index \
--genomeFastaFiles genome/NC_000021.9.fna \
--sjdbGTFfile annotation/NC_000021.9.gtf \
--runThreadN 2
STAR --runMode genomeGenerate --genomeDir genome/star_index/ --genomeSAindexNbases 7 --genomeFastaFiles genome/NC_000932.1.fasta --sjdbGTFfile genome/NC_000932.1.gtf --runThreadN 2
Alinear el genoma
| Parámetro | Descripción |
|---|---|
--readFilesIn |
archivo de reads a mapear |
--genomeDir |
indica donde esta alojado el genoma |
--runThreadN |
cantidad de hebras |
--outSAMType |
tipo de archivo SAM o BAM en este caso indicamos BAM |
SortedByCoordinate |
Ordenar el archivo BAM por las coordenadas del genoma |
--outFileNamePrefix |
Prefijo para los archivos de salida |
Command
# Help
STAR -h
# Run STAR (~3min)
STAR \
--genomeDir genome/star_index \
--readFilesIn results/trimmed/sample_filtered.fq \
--runThreadN 2 \
--outSAMtype BAM SortedByCoordinate \
--quantMode GeneCounts
STAR --genomeDir ../../../genome/star_index/ --readFilesIn ../../trimmed/NC_000932.1_6_trimmed.fq --runThreadN 2 --outSAMtype BAM SortedByCoordinate --limitBAMsortRAM 12000000000 --quantMode GeneCounts
# Move the BAM file into the correct folder
mv -v results/4_aligned_sequences/sampleAligned.sortedByCoord.out.bam results/4_aligned_sequences/aligned_bam/
# Move the logs into the correct folder
mv -v results/4_aligned_sequences/${BN}Log.final.out results/4_aligned_sequences/aligned_logs/
mv -v results/4_aligned_sequences/sample*Log.out results/4_aligned_sequences/aligned_logs/
Output
── results/map/
└── aligned_bam/sampleAligned.sortedByCoord.out.bam <- Sorted BAM alignment fole
└── aligned_logs/sampleLog.final.out <- Log of STAR alignment rate
└── aligned_logs/sampleLog.out <- Log of steps take during STAR alignment
CONTEO DE SECUENCIAS
featureCounts -h
# guardar los archivos en una variable
dirlist=$(ls -t ./*.bam | tr '\n' ' ')
echo $dirlist
# Correr featureCounts de todas las muestras (~4 minutes)
featureCounts \
-a ../../annotation/* \
-o ../../results/5_final_counts/final_counts.txt \
-g 'gene_name' \
-T 4 \
$dirlist
Output
── results/counts/
└── final_counts.txt <- Final gene counts across all samples
└── final_counts.txt.summary <- Summary of gene summarization
```bash
# Se puede utilzar el program HTSEQ para el mismo proposito
htseq-count -s no -r pos -t exon -f bam <BAMFILE> <GTF> > <SALIDA>
IMPORTAR A R
library(DESeq2)
library(ggplot2)
library(clusterProfiler)
library(biomaRt)
library(ReactomePA)
library(DOSE)
library(KEGG.db)
library(org.Hs.eg.db)
library(pheatmap)
library(genefilter)
library(RColorBrewer)
library(GO.db)
library(topGO)
library(dplyr)
library(gage)
library(ggsci)
data <- read.table("final_counts.txt", header = TRUE, skip = 1, row.names = 1)
#head(data)
data <- data[,c(-1:-5)]
colnames(data) <- gsub("_21.bam", "", colnames(data), fixed = T)
colnames(data) <- gsub("_21.bam", "", colnames(data), fixed = T)
colnames(data) <- gsub("..", "", colnames(data), fixed = T)
#data
metadata <- read.delim("muestras.txt", row.names = 1)
metadata$sampleid <- row.names(metadata)
metadata <- metadata[match(colnames(data), metadata$sampleid),]
#head(metadata)
#condition <- factor(c("A2058","A375","C32","Malme3M","SKMEL28", "SKMEL5","WM2664"))
#design <- as.formula(~Group)
#modelMatrix <- model.matrix(design, data = metadata)
#modelMatrix
ddsMat <- DESeqDataSetFromMatrix(countData = data, colData = metadata , design = ~1)
ddsMat <- DESeq(ddsMat)
Warning message in DESeq(ddsMat):
“the design is ~ 1 (just an intercept). is this intended?
estimating size factor
estimating dispersion
gene-wise dispersion estimate
mean-dispersion relationshi
final dispersion estimate
fitting model and testin
-- replacing outliers and refitting for 18 gene
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds
estimating dispersion
fitting model and testi
results <- results(ddsMat, pAdjustMethod = "fdr", alpha = 0.05)
summary(results)
out of 432 with nonzero total read count
adjusted p-value < 0.05
LFC > 0 (up) : 225, 52%
LFC < 0 (down) : 0, 0%
outliers [1] : 0, 0%
low counts [2] : 10, 2.3%
(mean count < 0)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
mcols(results, user.names = True)
DataFrame with 6 rows and 2 columns
type description
<character> <character>
baseMean intermediate mean of normalized c..
log2FoldChange results log2 fold change (ML..
lfcSE results standard error: Inte..
stat results Wald statistic: Inte..
pvalue results Wald test p-value: I..
padj results fdr adjusted p-values
results$description <- mapIds( x = org.Hs.eg.db, keys = row.names(results), column = "GENENAME", keytype = "SYMBOL", multiVals = "first")
results$symbol <- row.names(results)
results$entrez <- mapIds(x = org.Hs.eg.db, keys= row.names(results), column = "ENTREZID", keytype = "SYMBOL", multiVals = "first")
results$ensembl <- mapIds(x = org.Hs.eg.db, keys = row.names(results), column = "ENSEMBL", keytype = "SYMBOL", multiVals = "first")
results_sig <- subset(results, padj < 0.05)
head(results_sig)
log2 fold change (MLE): Intercept
Wald test p-value: Intercept
DataFrame with 6 rows and 10 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
GATD3B 36.51642 5.19047 0.272218 19.06737 4.71426e-81
LOC102724159 50.25903 5.65131 0.327048 17.27974 6.68489e-67
LOC102724200 8.13856 3.02477 0.480640 6.29322 3.10953e-10
SIK1B 19.51849 4.28677 0.362775 11.81662 3.20333e-32
LOC102724701 36.76702 5.20034 0.338991 15.34063 4.09180e-53
CBSL 16.40618 4.03617 0.617085 6.54070 6.12330e-11
padj description symbol entrez
<numeric> <character> <character> <character>
GATD3B 1.96972e-80 glutamine amidotrans.. GATD3B 102724023
LOC102724159 2.66134e-66 periodic tryptophan .. LOC102724159 102724159
LOC102724200 8.00135e-10 trafficking protein .. LOC102724200 102724200
SIK1B 1.11719e-31 salt inducible kinas.. SIK1B 102724428
LOC102724701 1.54173e-52 uncharacterized LOC1.. LOC102724701 102724701
CBSL 1.65643e-10 cystathionine beta-s.. CBSL 102724560
ensembl
<character>
GATD3B ENSG00000280071
LOC102724159 ENSG00000275464
LOC102724200 NA
SIK1B ENSG00000275993
LOC102724701 ENSG00000275496
CBSL ENSG00000274276
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
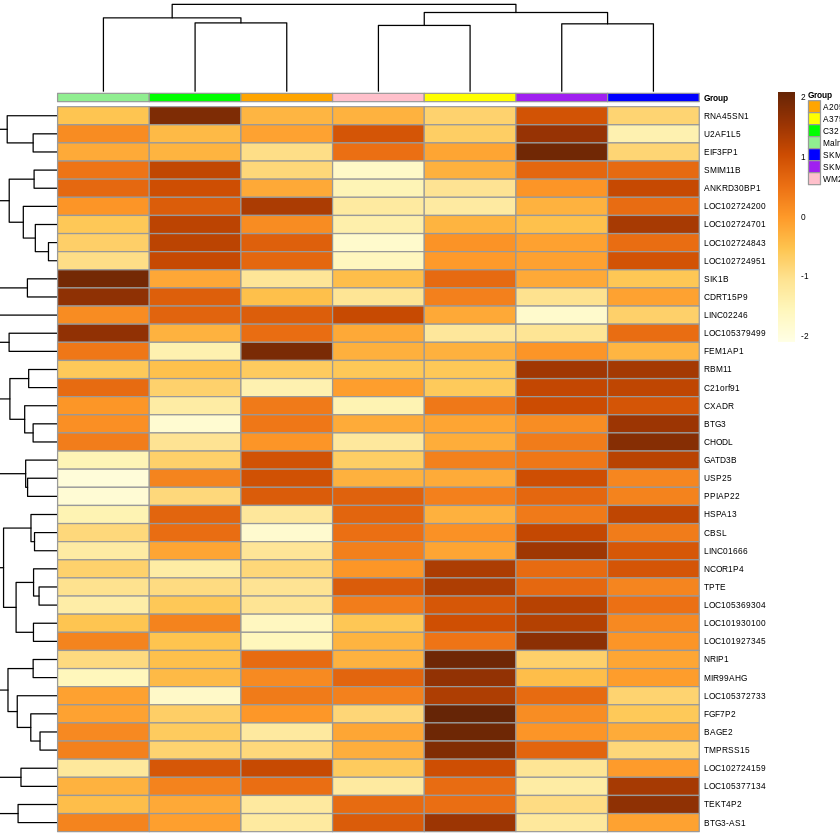
mat <- assay(ddsMat_rlog[row.names(results_sig)])[1:40, ]
##
annotation_col = data.frame(
Group = factor(colData(ddsMat_rlog)$Group),
row.names = colData(ddsMat_rlog)$sampleid
)
##
ann_colors = list(
Group = c(A2058 = "orange", A375 = "yellow", C32 = "green", Malme3M = "lightgreen", SKMEL28 = "blue", SKMEL5 = "purple", WM2664 = "pink"),
sampleid= c(a = "green")
)
##
pheatmap(mat = mat,
color = colorRampPalette(brewer.pal(9, "YlOrBr"))(255),
scale = "row", # Scale genes to Z-score (how many standard deviations)
annotation_col = annotation_col, # Add multiple annotations to the samples
annotation_colors = ann_colors,# Change the default colors of the annotations
fontsize = 5, # Make fonts smaller
cellwidth = 55, # Make the cells wider
show_colnames = F)