Indice del Curso »Siguiente Pagina

Introducción a Bioinformática
CONTENIDO

BASH PIPELINES
COMANDO PRÁCTICOS
Eliminar espacios en blanco
sed '/^$/d' file.txt
grep . file.txt
grep "\S" file.txt
Imprimir lineas entre rangos
awk 'NR>=20&&NR<=80' input.txt
Convertir fastq a fasta
seqtk seq -a fastq_file.fq > fasta_file.fa
Separar multifasta
awk '/^>/{s=++d".fa"} {print > s}' multi.fa
Separar una secuencia especifica de un fasta
samtools faidx file.fasta
samtools faidx file.fasta "ID_seq" > ID_seq.fasta

PYTHON SCRIPTS
Instalando Packages
Instalación de PIP
sudo apt-get install python3 python3-pip
pip3 install biopython
pip3 install Bio
Instalación conda Miniconda Anaconda
bash <Mini/A>conda3-latest-Linux-x86_64.sh
conda install -c conda-forge biopython
Importando Packages
from Bio import Entrez
Entrez.email = "francisco.ascue@unmsm.edu.pe"
handle = Entrez.efetch(db="Nucleotide", id="AY994334.1",rettype="fasta",retmode="text")
print(handle.read())
sudo pip3 install dna_features_viewer
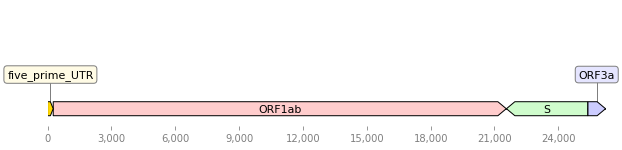
from dna_features_viewer import GraphicFeature, GraphicRecord
features=[
GraphicFeature(start=0, end=265, strand=+1, color="#ffd700",
label="five_prime_UTR"),
GraphicFeature(start=266, end=21555, strand=+1, color="#ffcccc",
label="ORF1ab"),
GraphicFeature(start=21563, end=25384, strand=-1, color="#cffccc",
label="S"),
GraphicFeature(start=25393, end=26220, strand=+1, color="#ccccff",
label="ORF3a")
]
record = GraphicRecord(sequence_length=26220, features=features)
ax, _ = record.plot(figure_width=10)
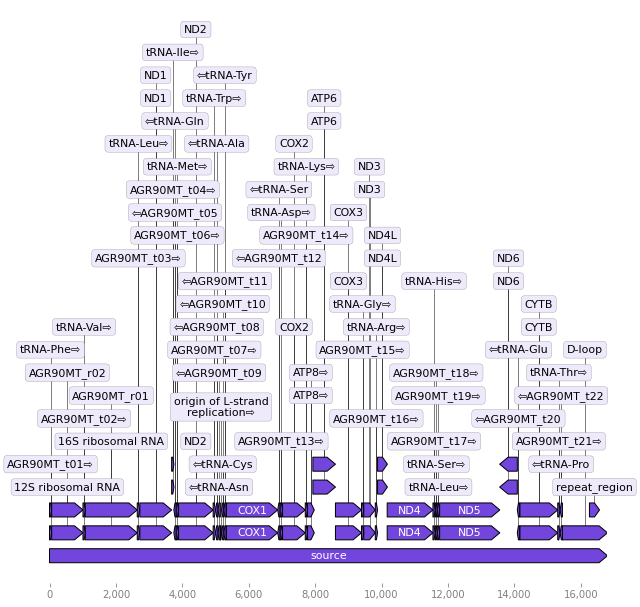
# import packages
from Bio import Entrez, SeqIO
from dna_features_viewer import annotate_biopython_record
from dna_features_viewer import BiopythonTranslator
# Download sequence in NCBI format
Entrez.email = "francisco.ascue@unmsm.edu.pe"
handle = Entrez.efetch( db="nucleotide", id="NC_000884.1", rettype="gb", retmode="text")
record = SeqIO.read(handle, "genbank")
#record.features = [ f for f in record.features if f.type not in ["gene"] ]
# Save records in to variale
SeqIO.write(record, "seq.gb", "genbank")
# Graphics
graphic_record = BiopythonTranslator().translate_record("seq.gb")
ax, _ = graphic_record.plot(figure_width=10, strand_in_label_threshold=7)
![]()
R SCRIPTS
Instalando Packages
install.package("BiocManager")
##Cargando packages
library(BiocManager)
BiocManager::install("ggplot2")
##Actualizando packages
install.packages(c("BiocManager","ggplot2")
update.packages(ask = FALSE)
### Cargar tablas (csv, tsv)
read.table("datafile.ext", sep = ",", header = TRUE)
read.csv("datafile.csv")
### Cargar paletas de colores
library(viridisLite)
library(viridis)
library(RColorBrewer)
display.brewer.all()

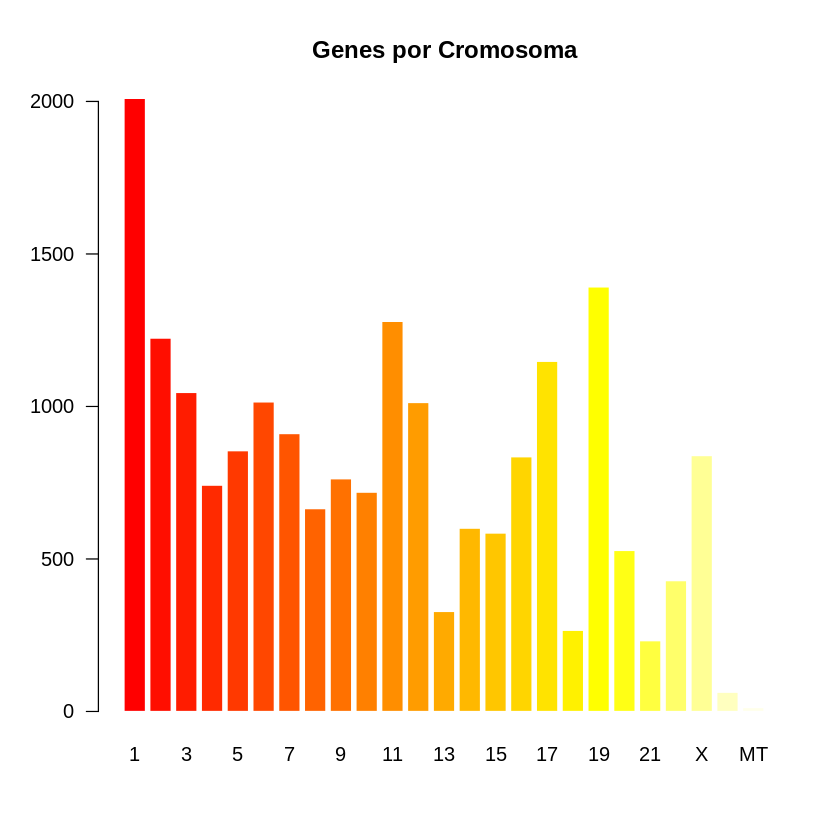
### barplot basicos
barplot(height = gene$V1, border = FALSE, names.arg = gene$V2,
las = 1 ,
col = heat.colors(25),
main = "Genes por Cromosoma")
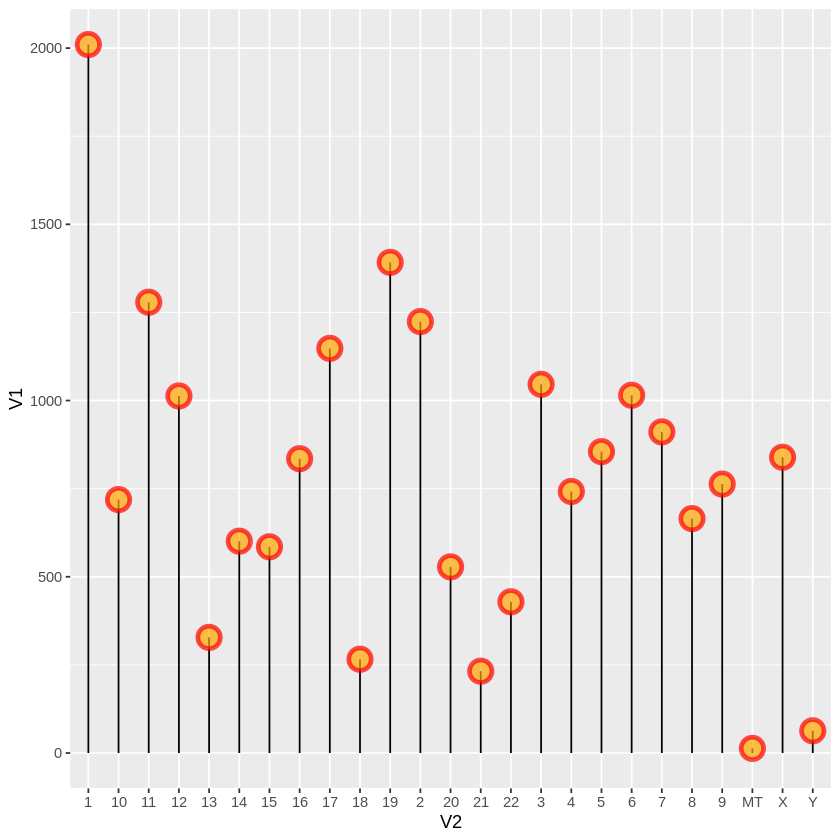
ggplot(gene, aes(x=V2, y=V1)) +
geom_segment( aes(x=V2, xend=V2, y=0, yend=V1)) +
geom_point( size=5, color="red", fill=alpha("orange", 0.3), alpha=0.7, shape=21, stroke=2)
ENTREZ NCBI PERL
esearch -db nucleotide -query "covid19"
<ENTREZ_DIRECT>
<Db>nucleotide</Db>
<WebEnv>MCID_606a64fe45460140b56b5fe4</WebEnv>
<QueryKey>1</QueryKey>
<Count>139219</Count>
<Step>1</Step>
</ENTREZ_DIRECT>
esearch -db nucleotide -query '("Severe acute respiratory syndrome coronavirus 2"[Organism] AND (viruses[filter])'
<ENTREZ_DIRECT>
<Db>nucleotide</Db>
<WebEnv>MCID_606a6ccaa53f343d4860d16b</WebEnv>
<QueryKey>1</QueryKey>
<Count>130415</Count>
<Step>1</Step>
</ENTREZ_DIRECT>
esearch -db nucleotide -query '("Severe acute respiratory syndrome coronavirus 2"[Organism] AND (viruses[filter])' -mindate 2021/01/01 -maxdate 2021/01/02
<ENTREZ_DIRECT>
<Db>nucleotide</Db>
<WebEnv>MCID_606a6f2559c60d0d1e6a4984</WebEnv>
<QueryKey>1</QueryKey>
<Count>59</Count>
<Step>1</Step>
</ENTREZ_DIRECT>
esearch -db nucleotide -query '("Severe acute respiratory syndrome coronavirus 2"[Organism] AND (viruses[filter])' -mindate 2020/01/01 -maxdate 2021/03/02 | efilter -country peru
<ENTREZ_DIRECT>
<Db>nucleotide</Db>
<WebEnv>MCID_606a6fb2fdbe9156e53f0caa</WebEnv>
<QueryKey>2</QueryKey>
<Count>90</Count>
<Step>2</Step>
</ENTREZ_DIRECT>
esearch -db nucleotide -query '("Severe acute respiratory syndrome coronavirus 2"[Organism] AND (viruses[filter])' -mindate 2021/01/01 -maxdate 2021/03/02 | efilter -country peru | efetch -format fasta
>MW494424.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/PER/LIM-INS-173/2020, complete genome
CTCGTCTATCTTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCAGCCGATCATCAGCACATCTAGGTTT
TGTCCGGGTGTGACCGAAAGGTAAGATGGAGAGCCTTGTCCCTGGTTTCAACGAGAAAACACACGTCCAA
CTCAGTTTGCCTGTTTTACAGGTTCGCGACGTGCTCGTACGTGGCTTTGGAGACTCCGTGGAGGAGGTCT
TATCAGAGGCACGTCAACATCTTAAAGATGGCACTTGTGGCTTAGTAGAAGTTGAAAAAGGCGTTTTGCC
TCAACTTGAACAGCCCTATGTGTTCATCAAACGTTCGGATGCTCGAACTGCACCTCATGGTCATGTTATG
GTTGAGCTGGTAGCAGAACTCGAAGGCATTCAGTACGGTCGTAG ...
esearch -db pubmed -query "Severe acute respiratory syndrome coronavirus2" | elink -related | efetch -format docsum | xtract -pattern Author -element Name | sort-uniq-count-rank
37 Li Y
26 Wang Y
25 Li J
24 Zhang J
24 Zhang Y
20 Wang J
20 Zhang S
19 Zhang L
18 Wang X
18 Zhang X
. .
. .
. .